![]()
![]()
![]()
![]()
Overview
This library performs unsupervised batch correction of high-dimensional data via the use of mutual nearest neighbors (MNNs). MNN correction was initially described in the context of single-cell RNA sequencing data analysis (see Haghverdi et al., 2018) but the same methodology can be applied for any high-dimensional data containing shared populations across multiple batches. This implementation is loosely based on the fastMNN() function in the batchelor package, which provides a number of improvements and speed-ups over the original method in the Haghverdi paper.
Quick start
Consider a dense matrix in column-major format where rows are dimensions (e.g., principal components) and cells are columns, and a vector of integers specifying the batch of origin for each cell. These are passed to mnncorrect::compute() to compute corrected values:
We also support batches in separate arrays, storing the corrected values for all batches in a single output array:
Advanced users can also fiddle with the options:
See the reference documentation for more details.
Theoretical details
We assume that (i) our batches share some subpopulations, and (ii) even after the addition of an arbitrary batch effect, the cells from one subpopulation in one batch are still closer to the cells in the corresponding subpopulation in the other batch (and vice versa) when compared to cells in different subpopulations. Thus, by identifying pairs of cells that are MNNs, we can determine which subpopulations are shared across batches. Any differences in location between batches for the shared subpopulations are attributed to batch effects and targeted for removal. In contrast, a subpopulation unique to a single batch will not contain any MNNs (and thus will not interfere with correction), as it will not have a corresponding subpopulation in the other batch for which it can be the closest neighbor.
To remove batch effects, we consider one batch to be the "reference" and another to be the "target". Each MNN pair defines a correction vector that moves the target cell towards its paired reference cell. For each cell $i$ in the target batch, we identify the closest cell in the same batch that is part of a MNN pair (i.e., "MNN-involved cells"). We apply that pair's correction vector to $i$ to obtain its corrected coordinates. The use of the closest MNN-involved cell allows the correction to adjust to local variations in the magnitude and direction of the batch effect. If an MNN-involved cell in the target batch is part of multiple MNN pairs, we only use the correction vector of the pair with the shortest distance between its paired cells, for simplicity.
The correction vector for each MNN pair is not directly computed from its two paired cells. Rather, for each cell, we compute a "center of mass" using neighboring points from the same batch. Each center of mass is defined as the centroid of the set of $k$ nearest neighbors of each MNN-involved cell. This can be done recursively with the neighbors of those neighbors, etc., up to a user-specified recursion depth. The aim is to mitigate "kissing" effects where the correction only brings the surfaces of the batches into contact.
In the case of >2 batches, we define a merge order based on the batch size, variance, residual sum of squares, or the input order. For the first batch to be merged, we identify MNN pairs to all other batches at once. The subsequent correction effectively distributes the first batch's cells to all other batches. This process is repeated for all remaining batches until only one batch remains that contains all cells. By using all batches to identify MNN pairs at each step, we improve the chance of correctly matching subpopulations across batches, even if they are missing from certain batches.
Examples
The tests/R/examples directory contains a few examples using the C++ code on some real datasets (namely, single-cell RNA-seq datasets). To run these, install the package at tests/R/package (this also requires the scrapper package for the various preprocessing steps):
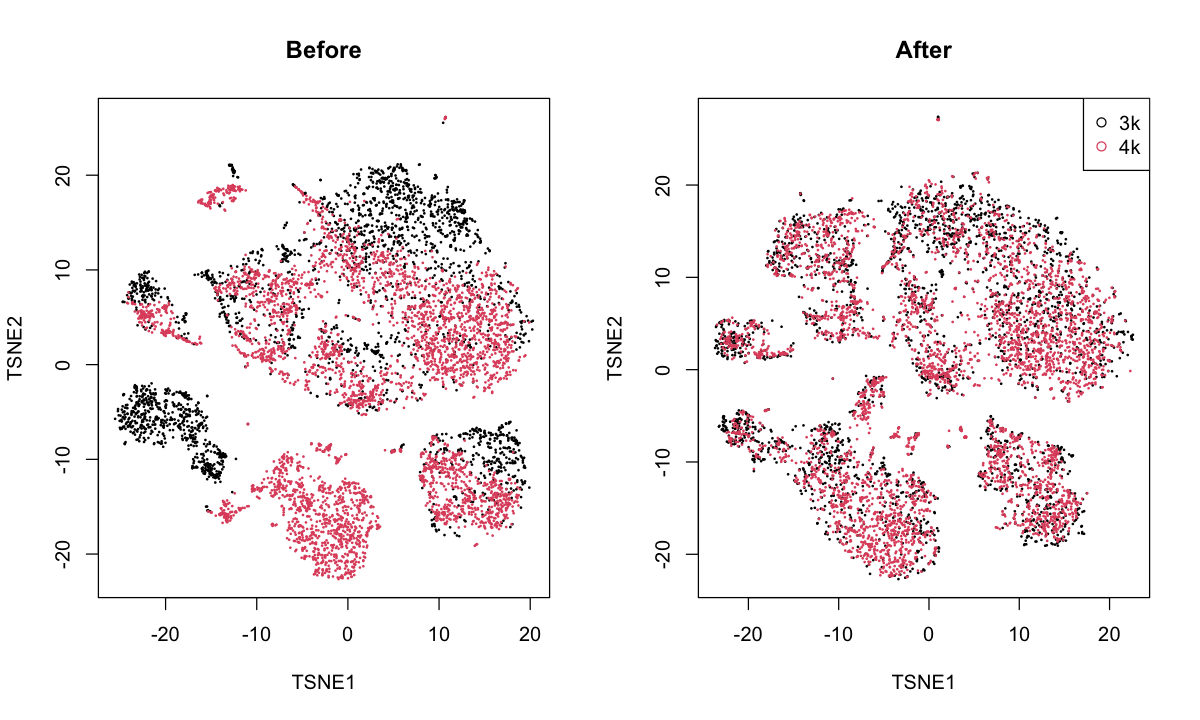
pbmc: merges the PBMC 3K and 4K datasets from 10X Genomics. These are technical replicates (I think) so a complete merge is to be expected.
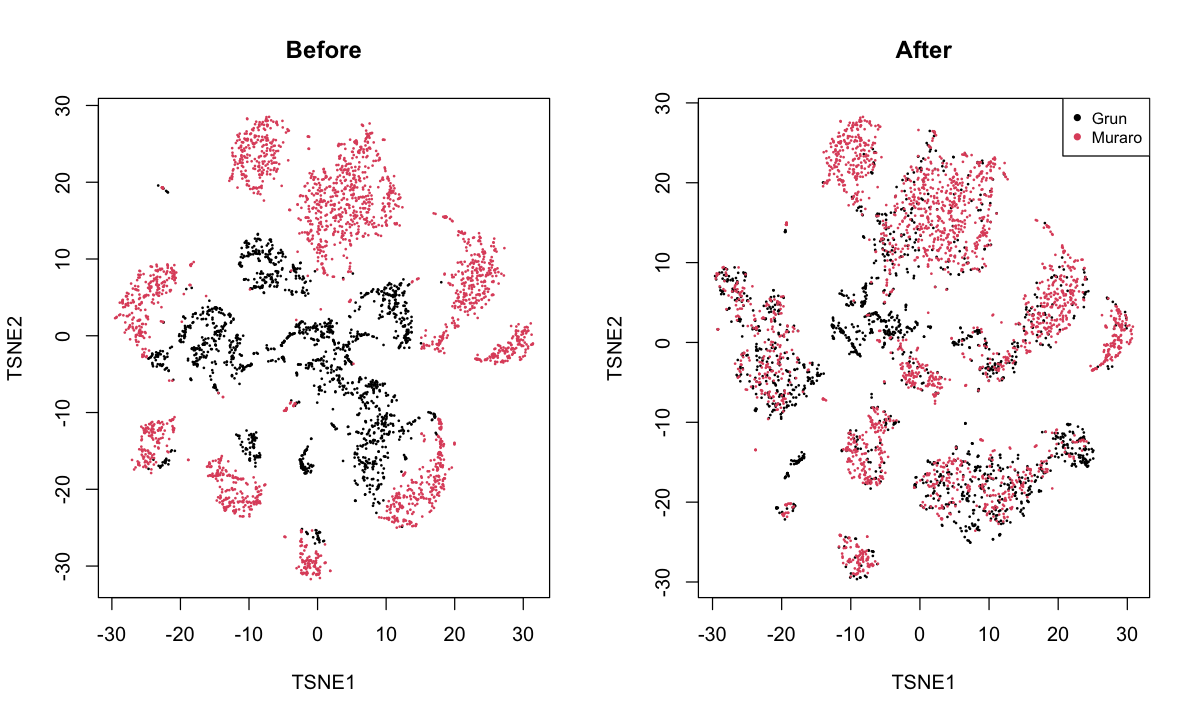
pancreas: merges the Grun et al. (2016) and Muraro et al. (2016) datasets. I believe this involves data from different patients but using the same-ish technology.
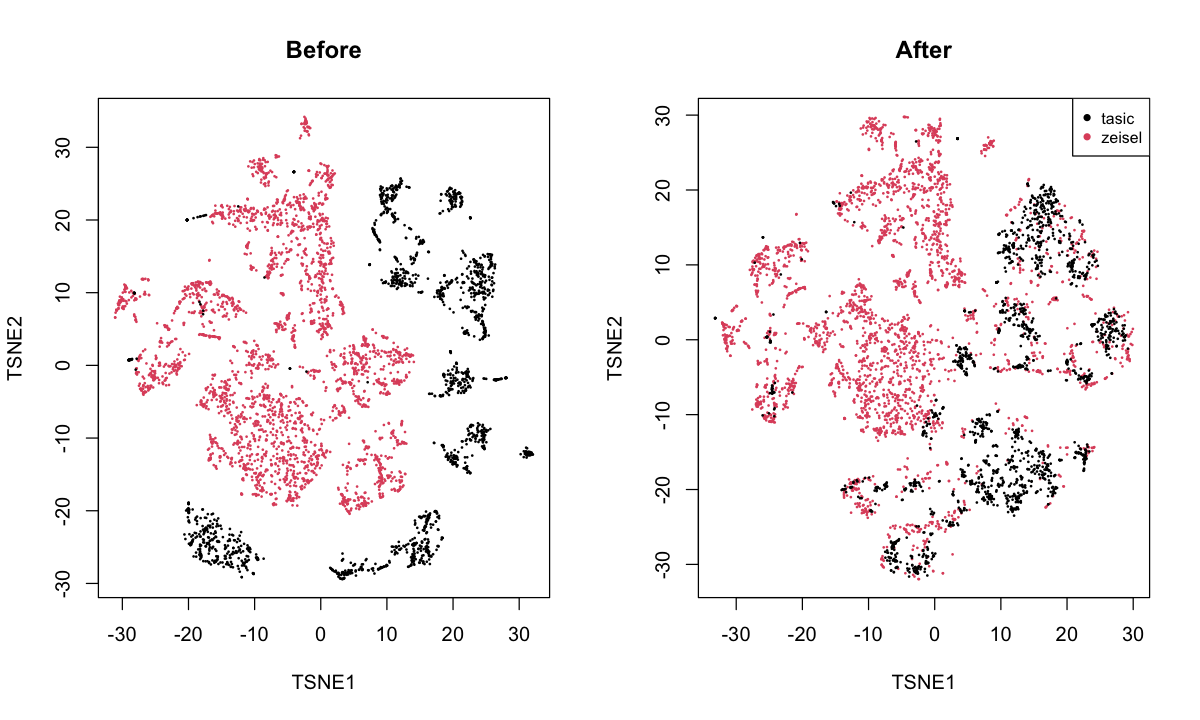
neurons: merges the Zeisel et al. (2015) and Tasic et al. (2016) datasets. This involves different technologies and different cell populations.
Building projects
CMake with FetchContent
If you're using CMake, you just need to add something like this to your CMakeLists.txt:
Then you can link to libscran to make the headers available during compilation:
By default, this will use FetchContent to fetch all external dependencies. Applications should consider pinning versions of dependencies for stability - see extern/CMakeLists.txt for suggested versions. If you want to install them manually, use -DMNNCORRECT_FETCH_EXTERN=OFF.
CMake with find_package()
To install the library, use:
Again, this will use FetchContent to retrieve dependencies, see comments above.
Manual
If you're not using CMake, the simple approach is to just copy the files - either directly or with Git submodules - and include their path during compilation with, e.g., GCC's -I. This also requires the external dependencies listed in extern/CMakeLists.txt.
References
Haghverdi L, Lun ATL, Morgan MD, Marioni JC (2018). Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36(5):421-427
Generated by